Marfan sendromu, bağ dokularını etkileyen genetik bir durumdur. Kalp, kan damarları, akciğerler, deri, kemikler, eklemler ve gözler dahil olmak üzere insan vücudunun farklı bölümlerini etkileyebilir.
Bağ dokuları, diğer doku ve organlara destek sağlamaya yardımcı olan karmaşık yapılardır.
Marfan sendromunun etkileri hafif ila yaşamı tehdit eder. En ciddi komplikasyonlar kalp kapakçığı, aort veya her ikisini de içerir. Bilişsel yetenek üzerinde hiçbir etkisi yoktur.
Bu, her 5000 kişide 1’i etkileyen kalıtsal bir durumdur.
Tedavi yoktur, ancak terapi, bir kişinin yaşam kalitesini iyileştirebilir ve olası tehlikeli komplikasyonları önleyebilir. Zamanında tedavi, Marfan sendromu olan bir kişinin, durumu olmayan bir bireyle aynı yaşam süresine sahip olduğu anlamına gelebilir.
Marfan sendromunda hızlı gerçekler
- Marfan sendromu çok çeşitli kalp, göz ve iskelet sorunlarına neden olabilen genetik bir durumdur.
- Semptomlar genellikle aortta olağan dışı uzun kollar ve parmaklar, gelişmiş boy ve gözyaşları içerir. Yetişkinliğe kadar farkedilemezler.
- Durum, bağ dokusunu güçlendiren bir gendeki sınırlamalardan kaynaklanır.
- Marfan sendromunun tedavisi yoktur, ancak belirtiler yönetilebilir ve rahatlatılabilir.
- Marfan sendromlu bir kişi, doğru koruyucu önlemler alındığında normal bir yaşam beklentisine sahip olabilir.
belirtiler
Marfan sendromu vücudun çeşitli bölgelerini etkileyebileceğinden, belirtiler insanlar arasında büyük farklılıklar gösterebilir.
Bununla birlikte, Marfan sendromu olan kişiler genellikle benzersiz bir fiziksel yapıya sahiptir.
Genellikle aortta gözyaşı ve genişleme veya kalpten önde gelen büyük kan damarı yaşarlar.
Marfan sendromlu farklı kişiler farklı semptomlar göstereceklerdir.
Semptomlar iskelet, göz ve kardiyovasküler sistemde ortaya çıkabilir. Bazı kişilerde, semptomlar hafiftir ve belirli vücut kısımlarıyla sınırlıdır. Diğerlerinde, bunlar şiddetlidir ve çeşitli vücut kısımlarını etkiler. Marfan sendromunun semptomları yaşla birlikte kötüleşmektedir.
İskelet sisteminde ortaya çıkabilen belirtiler ve semptomlar şunlardır:
- ince ve zayıf bileklere sahip uzun uzuvlar
- omuzları
- çok uzun ve ince parmaklar ve ayak parmakları veya her ikisi de
- sternum veya göğüs kemiği, içinde çıkıntı veya mağaralar
- son derece esnek eklemler
- uzun ve dar yüz
- konuşma bozukluklarına neden olabilen küçük alt çene
- aşırı kalabalık dişler
- ince gövde ve ortalama boydan uzun
- düz ayak
- konuşma bozukluklarına neden olabilecek yüksek damak
- Hamilelik veya kilo alımından dolayı ciltte gerilmeler
- eklemlerde, kemiklerde ve kaslarda ağrı
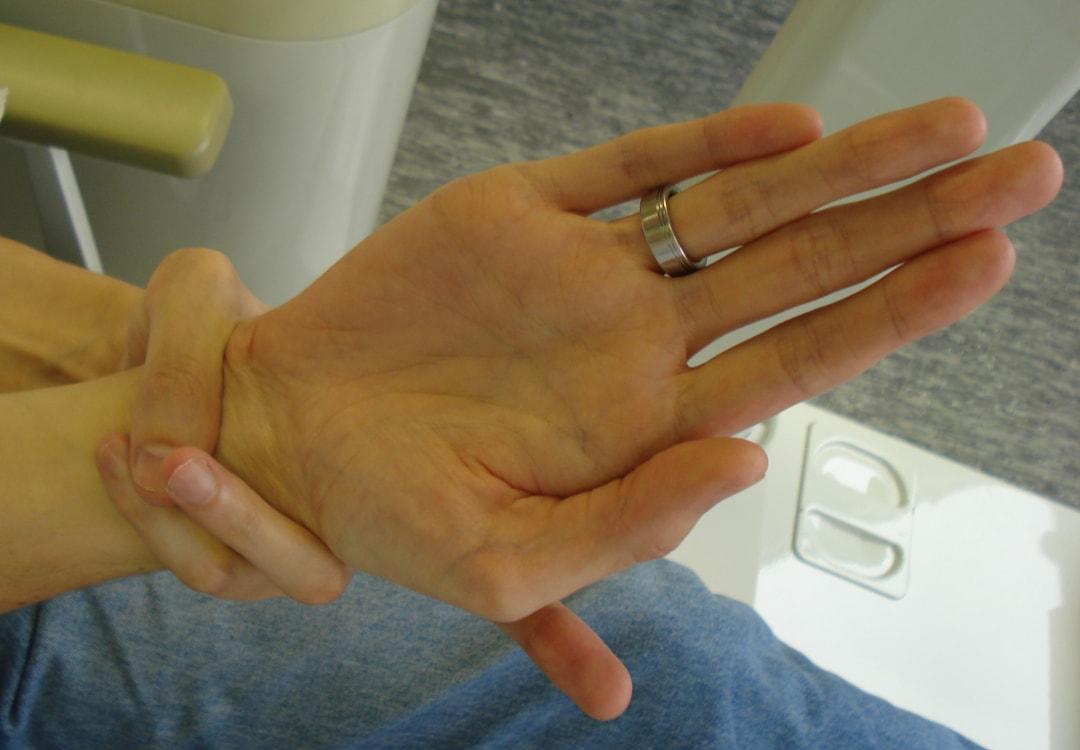
Marfan sendromuna özgü uzun uzuvlar genellikle bireyin kol açıklığının boyundan daha uzun olduğu anlamına gelir.
Skolyoz veya omurganın eğriliği, spondilolistez ve dural ektazinin gelişme riski daha yüksektir. Dural ektazi omuriliği çevreleyen dural kesenin genişlemesi veya balonlanmasıdır.
Gözlerle ilgili problemler şunlardır:
- miyopluk
- astigmatizm
- mercek dislokasyonu
- müstakil retina
Marfan sendromu da erken başlangıçlı glokom ve kataraktlara yol açabilir.
Marfan sendromunda aort düzensizlikleri aşağıdaki kalp sorunlarına yol açabilir:
- yorgunluk
- nefes darlığı
- çarpıntı veya kalp üfürüm
- angina, göğüste ağrı ile sırt, omuz ya da kol yayılıyor
- aort da dahil olmak üzere kalp kapaklarının prolapsusu
- dilate aort
- aort anevrizması
Marfan sendromu belirtileri yaşamın ilerleyen dönemlerine kadar ortaya çıkmayabilir.
Nedenler
 Marfan sendromu genetik bir durumdur. Ya ailelerden geçilir ya da bir sperm ya da yumurtadaki rastlantısal olarak ortaya çıkan kusurlu bir gen nedeniyle olur.
Marfan sendromu genetik bir durumdur. Ya ailelerden geçilir ya da bir sperm ya da yumurtadaki rastlantısal olarak ortaya çıkan kusurlu bir gen nedeniyle olur.
Bu sendromu olan çoğu insan bir ebeveynden miras alır. Bu durumdaki bir ebeveyni çocuğa geçirme şansı yüzde 50’dir, ancak hastaların yüzde 25’inde bu durumun ebeveynleri yoktur. Bu spontan mutasyon olarak bilinir.
Marfan sendromunu taşıyan gen FBN1 olarak adlandırılır. FBN1, esnek doku ve mukavemet ile bağ dokusu sağlayan Fibrillin-1 adlı bir protein sağlar.
Bağ dokudaki bu nitelikler olmadan, vücudun hareket etmesi ve bükülmesi zordur. Bağ dokusu, organları ve çeşitli vücut kısımlarını destekleyemez hale gelir.
Marfan’daki kusurlu gen, Fibrillin-1 üretimini durdurur ve sitokin olarak adlandırılan ve daha sonra inflamasyona ve skarlaşmaya yol açabilen bir protein seviyesinin artmasına neden olur.
Teşhis
Marfan sendromunun tanısı, semptomların aralığı göz önüne alındığında zor olabilir.
Hekim:
- Hastanın ailesini ve tıbbi geçmişini sormak
- hastanın kalbini dinle
- cilt çatlakları için kontrol edin
- hastanın kolları, bacakları, parmakları ve ayak parmaklarının uzunluğuna ve özelliklerine bakın
Ehlers-Danlos sendromu veya Beals sendromu gibi üst üste gelen semptomlarla birlikte diğer koşulların da göz ardı edilmesi gerekir.
Doktor, bir çocuk genç oluncaya kadar tanı koymayabilir. İşaretler ve semptomlar daha net olacaktır.
Bir doktor, tanıyı doğrulamak için aşağıdaki testleri kullanabilir:
- kalp, kapak ve aort durumunu kontrol etmek için ekokardiyogram
- kalp hızı ve ritmi kontrol etmek için elektrokardiyogram (EKG)
- yarık lamba göz muayenesi, çıkık lensleri kontrol etmek için
- Dural ektazinin bulguları için alt sırtını kontrol etmek için BT veya MRI taramaları kullanılabilir.
- genetik test bazen tavsiye edilebilir
tedavi
Marfan sendromu için tedavi yoktur. Bununla birlikte, tedavi semptomları hafifletebilir ve olası komplikasyonları en aza indirebilir veya önleyebilir.
Doktor kişiselleştirilmiş bir tedavi programı geliştirecektir. Detaylar etkilenen vücut parçalarına ve sistemlere bağlı olacaktır.
Düzenli izleme, omurga ya da sternumdaki değişiklikleri erken bir aşamada tanıyarak kemik, eklem ve doku problemlerini önlemeye yardımcı olabilir. Bu özellikle Marfan sendromlu bir çocuk bir çocuk ise ve hala büyüyorsa faydalı olabilir.
Gecikmiş tedavi, kalp ve akciğer fonksiyonlarını zayıflatabilir. Ortopedik bir ayraç veya hatta ameliyat gerekebilir.
Düzenli göz kontrolleri, herhangi bir görme probleminin derhal tespit edilmesini ve tedavi edilmesini sağlayabilir. Ulusal Marfan Vakfı’na göre, çoğu görme problemi gözlük ve lenslerle düzeltilebilir. Bazı hastalarda düzeltici ameliyat gerekebilir.
Kan damarları ve kalbin düzenli izlenmesi gerekecektir. Kalp ve aort problemlerinin erken tespiti ve tedavisi komplikasyonları önleyebilir.
Beta-blokerler kalp kapakçık problemleri için ve aort içindeki basıncı azaltmak için reçete edilebilir. Bazen aort tamir etmek veya bir kalp kapakçık değiştirmek için ameliyat gereklidir. Acil cerrahi girişim olası aort diseksiyonuna, aort duvarında ciddi bir yırtılma veya yırtılmaya karşı önemli bir koruyucu önlemdir.
Marfan Vakfı, kalp problemi olan hastalara bir medikal uyarı bileziği takmaları ve herhangi bir göğüs, sırt ya da karın ağrısı hissetmeleri durumunda doğrudan hastaneye gitmelerini önerir.
Marfan sendromu ile ortaya çıkabilen dural ektaziler, merkezi sinir sistemini (CNS) etkileyebilir. Bu durumlarda, hastanın acıyı tedavi etmek için ilaca ihtiyacı olabilir.
Görünüm
Marfan sendromu olan kişilerin yüksek yoğunluklu temas sporlarına katılmamaları tavsiye edilir. Bowling veya golf gibi fiziksel çarpışmalara veya yoğun çaba gerektirmeyen sporlara izin verilir.
Bir sporcuya hangi sporların uygun ve güvenli olduğunu sormak önemlidir.
Marfan Vakfı’nın Baş Bilim Müdürü olan Josephine Grima şunları söyledi:
“Marfan sendromunun temel anlayışının ve tedavisinin ilerlemesinde büyük ilerleme kaydedilmiştir. Sonuç olarak, Marfan ve ilgili hastalıkları olan kişiler teşhis edilirse ve uygun tıbbi tedavi almaları halinde normal bir yaşam sürdürebilirler.”
Araştırma hakkında yardım, destek ve bilgi için Marfan Vakfı web sitesini ziyaret edin.